Browse categories
Explore
Fiverr Pro
English
$
USD
About This Service
Are you working on a protein-ligand system and need reliable, well-analyzed molecular dynamics simulations for your research or publication? I provide end-to-end AMBER MD simulation services from system preparation to publication-ready figures using the same pipeline used in peer-reviewed computational drug discovery research.
I am a computational biology researcher with hands-on experience in AMBER/AmberTools, cpptraj, and Python-based analysis workflows. My work has directly supported manuscripts submitted to peer-reviewed journals in computational pharmacology.
What I Deliver
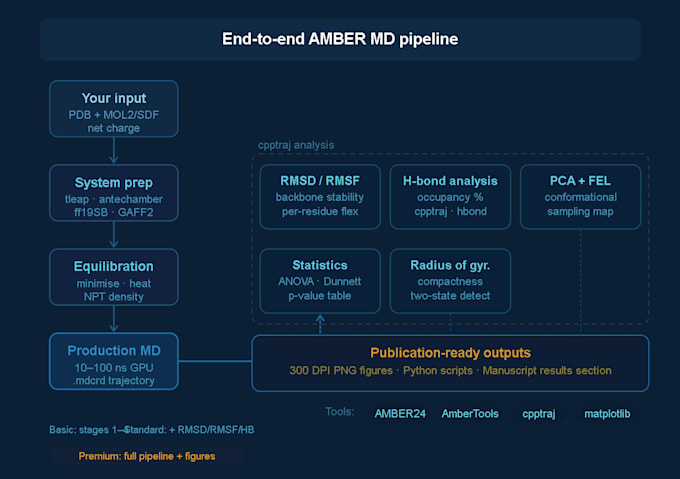
System Setup
Protein structure preparation and protonation state assignment
Ligand parameterization using antechamber
Solvation (TIP3P or OPC water box), counterion addition
Force field assignment: ff19SB (protein), GAFF2 (small molecule)
Minimization, heating, and NPT/NVT equilibration
Production MD Run
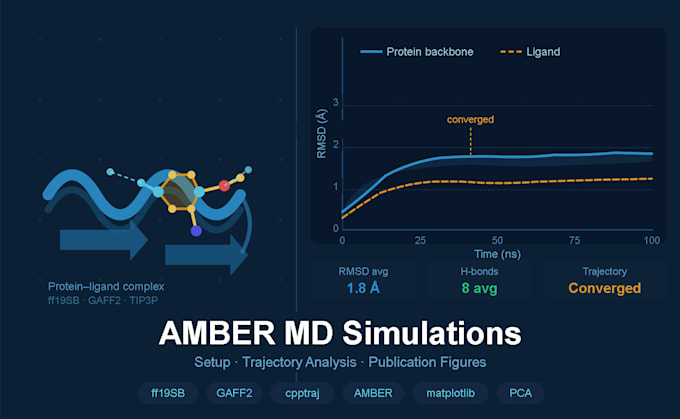
10, 50, or 100 ns production trajectories (package-dependent)
Full .mdcrd trajectory files delivered
Trajectory Analysis (Standard & Premium)
Full publications ready figures
Why Work With Me?
Real amber experience, not a cloud wrapper or online tool.